GABRIELA ESPIN O., ALICIA TORRES M., JESUS CASTRO V., RODOLFO BERNAL C.
Tipo:
Case Report
ABSTRACT (English):
Introduction: McCune-Albright syndrome (MAS) is a rare and complex genetic disease that affects the skin, skeleton, and endocrine system. The clinical triad described is composed of polyostotic fibrous bone dysplasia, café-au-lait skin pigmentation, and precocious puberty. When it is associated with myxoma, it is called Mazabraud's syndrome. The variable constellation of symptoms arises from a somatic activating mutation of the GNAS gene, which is present in many types of tissues.
Clinical Case: The case of a 6-year-old patient is presented, with a clinical presentation of a bone lesion in the left superciliary region with progressive growth for 10 months, suggestive of MAS. We focus on the course of the disease and neurosurgical treatment with the Neuronavigation technique, all carried out in our institution.
Conclusion: McCune Albright syndrome is an example of the genetic heterogeneity of polyostotic fibrous dysplasia, it is benign and has a low prevalence, with a great impact on quality of life, therefore treatment must be timely and multidisciplinary.
Keywords: Fibrous Dysplasia, Polyostotic, Puberty, Precocious, Craniofacial Fibrous Dysplasia (Source: MeSH NLM)
ABSTRACT (Spanish):
Introducción: El síndrome de McCune-Albright (SMA) es una rara y compleja enfermedad genética afecta a la piel, el esqueleto y el sistema endocrino. La tríada clínica descrita está compuesta por displasia fibrosa poliostótica del hueso, pigmentación de la piel café con leche y pubertad precoz. Cuando se asocia a mixoma se denomina síndrome de Mazabraud. La constelación variable de síntomas surge de una mutación activadora somática del gen GNAS, que está presente en muchos tipos de tejidos.
Caso Clínico: Se presenta el caso de una paciente de 6 años, con presentación clínica de una lesión ósea en la región superciliar izquierda con crecimiento progresivo desde hace 10 meses, sugestiva de SMA. Nos enfocamos en el curso de la enfermedad y tratamiento neuroquirúrgico con técnica de neuronavegación, todo realizado en nuestra institución hospitalaria
Conclusión: El síndrome de McCune Albright es un ejemplo de la heterogeneidad genética de la displasia fibrosa poliostótica, es de carácter benigno y prevalencia baja, con gran impacto en la calidad de vida por lo que el tratamiento debe se oportuno y multidisciplinario.
Palabras Clave: Displasia Fibrosa Poliostótica, Pubertad Precoz, Displasia Fibrosa craneofacial (Fuente: DeCS Bireme)
INTRODUCTION
McCune-Albright syndrome (MAS) is a rare genetic disorder originally recognized by the triad of polyostotic fibrous dysplasia, precocious puberty, and café-au-lait spots. Its estimated prevalence is between 1 in 100,000 and 1 in 1,000,000. The syndrome is caused by somatic activation mutations of the GNAS1 gene, this mutation results in an
alteration of the biochemical pathways which ultimately leads to constitutive activation of G proteins and proliferation of undifferentiated mesenchymal cells. 1.2
Craniofacial injuries in McCune-Albright syndrome can present as facial deformities or asymmetries. In general, 98% of patients have fibrous dysplasia at the diagnosis. 1
The diagnosis of MAS was originally clinical, based on the triad described. However, other clinical definitions have now been presented, including the presence of bone involvement that accompanies any characteristic endocrine or skin disorder. 3
We present the case of a patient with McCune-Albright syndrome who underwent neurosurgical treatment with the Neuronavigation technique.
CLINICAL CASE
History and examination: 6-year-old female patient, referred by the Endocrinology Service, with a history of precocious puberty characterized by menarche at 3 years and Tanner stage 2-3, the chronological age of 4.4 years, and bone age of 7 years. She was evaluated by Neurosurgery where the diagnosis of a pituitary tumor was ruled out by means of tomography and brain MRI studies. The patient had a 10-month history of the disease, characterized by presenting a bone lesion at the left superciliary level that grew progressively after a mild craniofacial trauma, associated with another ipsilateral parietal lesion. On physical examination: No signs of neurological targeting or cranial nerve alteration. At the cranial level, two non-mobile, non-painful, rounded lesions of a hard consistency were evidenced; the first superciliary, 3 cm in diameter, which deformed the superciliary arch, and the second lesion at the ipsilateral parietal level, 1.5 cm. At the skin level, two café-au-lait lesions were seen at the lumbar level. Tanner's stage was 2 to 3.
The Neurosurgery Service requested a skull tomography with Neuronavigation protocol and based on the findings found in the 3D bone window, it was decided to hospitalize her for surgical planning after evaluation by Endocrinology and Maxillofacial Surgery. The endocrinological evaluation found an excess of growth hormone, adrenocorticotropic hormone, thyroid-stimulating hormone, and gonadotropin deficiencies, for which leuprolide acetate 3.75 mg was prescribed intramuscularly.
After evaluating the Neuronavigation protocol, several foci of fibrous dysplasia were evidenced: 1) at the facial level involving the frontal bone, roof, and external face of the orbit and left malar, 2) at the left parietal level. (Fig 1)
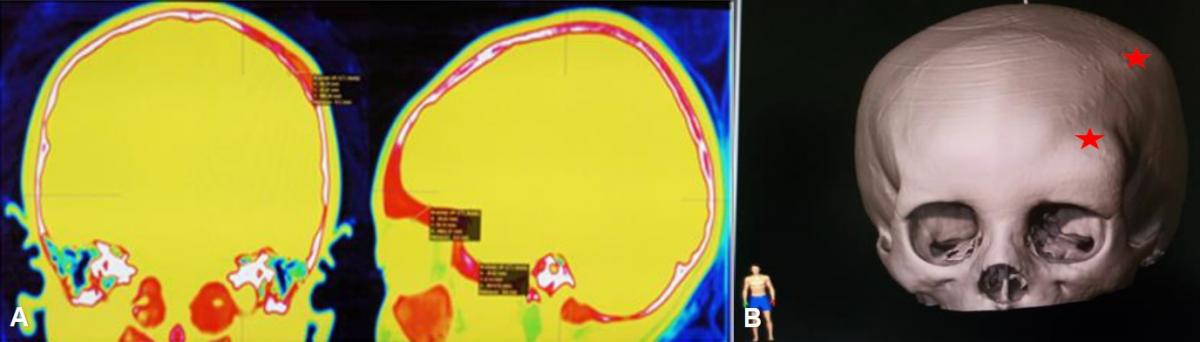 Fig 1. (A) Cranial tomography with navigation protocol. (B) Area of bone dysplasia at the left facial and left parietal level (red star). |
Surgical treatment: With these imaging findings, the total excision of the left parietal lesion and the placement of a titanium mesh cranioplasty were planned in the same surgical procedure. Maxillofacial surgery deferred the surgical procedure pending the histopathology result since the facial lesions were extensive.
A 6 cm left parietal craniectomy was performed with a 1 cm safety margin, 2 lesions of left parietal fibrous dysplasia were also evidenced, which were sent for histopathological study in a single parietal bone fragment. Subsequently, a titanium mesh was placed in the bone defect. (Fig. 2)
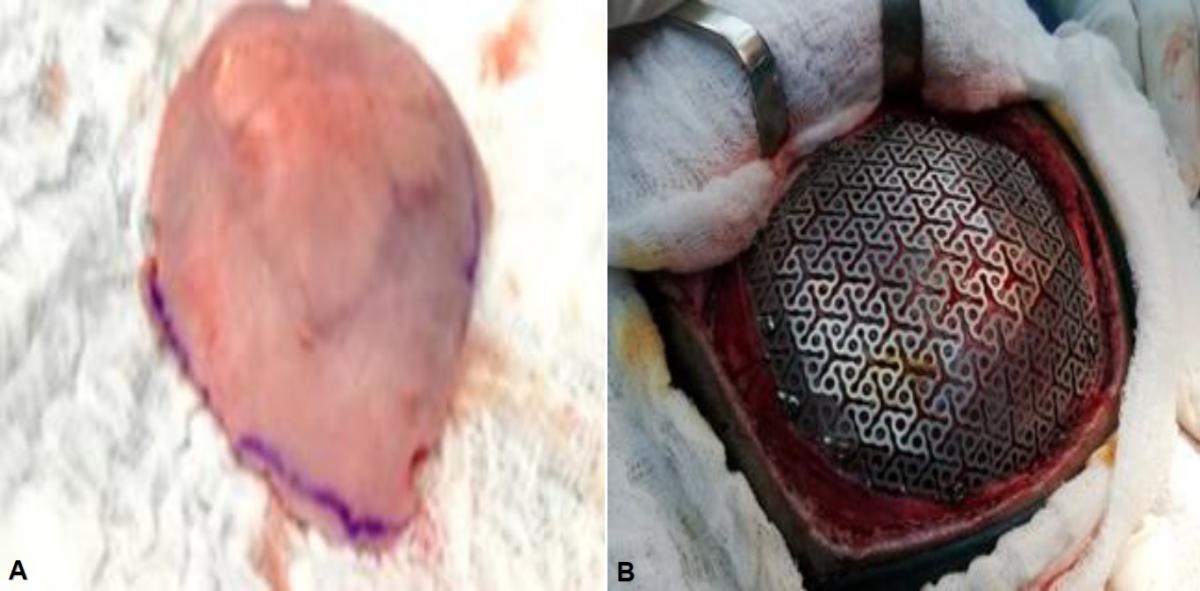 Fig 2. (A) Fibrous bone dysplasia in the left parietal bone, showing two lesions in the extracted bone platelet. (B) Cranioplasty with titanium mesh.
|
Postoperative evolution: The patient evolved favorably in the postoperative period. The histopathology result reported fibrous dysplasia formed by dense fibroblastic stroma with little cellularity and figures of newly formed trabecular bone, accompanied by small interstitial capillaries. No signs of cellular atypia. (Fig. 3).
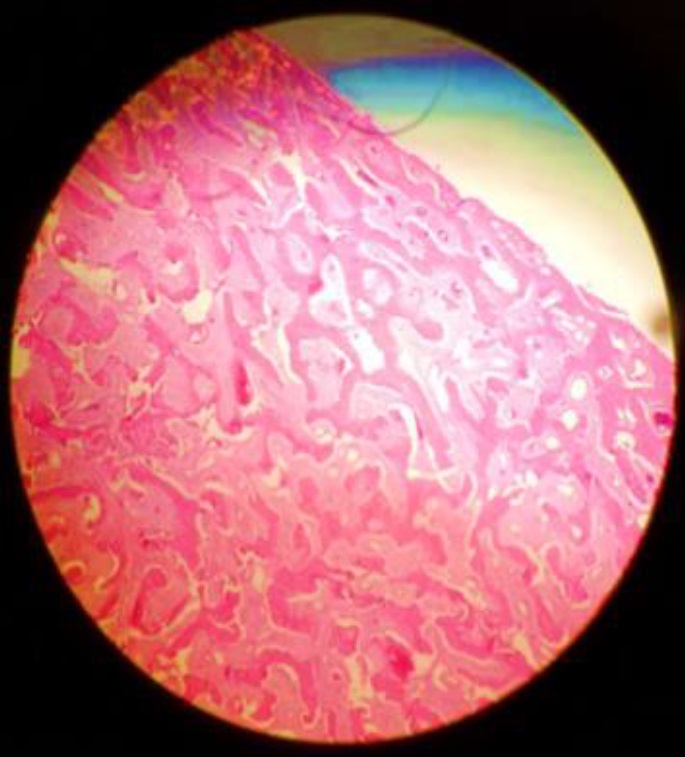 Fig 3. Histopathology image with fibroblastic stroma, newly formed trabecular bone compatible with fibrous dysplasia (H-E 10X)
|
DISCUSSION
The case of a 6-year-old patient with early menarche, café-au-lait spots, and bone lesions at the cranial level is reviewed and presented. In imaging studies, bone changes compatible with polyostotic dysplasia are observed and in association with the clinical context, the diagnosis of McCune-Albright syndrome (MAS) is made.
Polyostotic fibrous dysplasia is not a frequent entity in the exposed age group, although it can present at any age. Its importance lies in the presentation of a benign alteration of bone tissue, of low prevalence characterized by the replacement of bone by fibrous bone tissue. It is also more common in the female sex. 16, 7
Physiologically and pathologically, it is explained by the mutation of the GNAS1 gene that affects the ectoderm, mesoderm, and endoderm, causing alteration of melanocytes, osteoblasts, and endocrine glands, respectively.17, 18.
Its wide clinical spectrum includes precocious puberty characterized by breast growth or vaginal bleeding in girls and genital growth accompanied by precocious sexual behavior in boys, as well as ophthalmic, auditory, or skeletal manifestations of variable severity and endocrinological disorders, are what condition its diagnosis 5, 7,9,11,15,16.
The disease has several extra-skeletal manifestations that can range from thyroid gland involvement, frequent hyperthyroidism, hypophosphaturia due to loss of renal phosphate tissues, which is related to recurrent bone fractures at a younger age, and permanent bone pain. 11,19.
The diagnosis of this disease is based on the classical triad and by taking DNA from affected blood, skin, or tissue. 18
The approach for the treatment of patients with this pathology must be multidisciplinary and comprehensive since several clinical and surgical specialties intervene to alleviate the clinical manifestations of the patient. 8,10,14,16
This is how the thyroid echo and TSH blood dosage are important, likewise, the contribution of phosphate and vitamin D4 improve bone pain and fractures, it has been reported that the consequences in adulthood can be obesity, alteration of the metabolism of carbohydrates. carbon increased cardiovascular risk and blood pressure figures. Surgical treatment is related to diagnostic, aesthetic, orthopedic, and functional rehabilitation purposes. 19, 20
All the clinical manifestations described above have a psychosocial and emotional impact and a decrease in final height due to early closure of the growth plates, which requires timely and multifunctional treatment.
CONCLUSION
The presentation of McCune Albright Syndrome is an example of the genetic heterogeneity of this disease and the diversity of bone, skin, and endocrinological symptoms that can be identified.
Although this entity is benign and the prevalence is extremely low, the impact on quality of life and normal development of the individual incurs the need for early detection of this disease and requires multidisciplinary management for the treatment of symptoms in patients. the moment of its appearance.
REFERENCES
-
Couturier, A., Aumaître, O., Gilain, L., Jean, B., Mom, T., & André, M. (2017). Craniofacial fibrous dysplasia: A 10-case series. European Annals of Otorhinolaryngology, Head and Neck Diseases, 134(4), 229–235.doi:10.1016/ j.anorl.2017.02.004
-
Coulter, C., Richards, R., Peterson, D., & Collier, J. (2014). Parietal skull reconstruction using immediately peek cranioplasty following resection for craniofacial fibrous dysplasia. Journal of Plastic, Reconstructive & Aesthetic Surgery, 67(8), e208–e209.doi: 10.1016/j.bjps. 2014. 02.015
-
Collins, M. T., Singer, F. R., & Eugster, E. (2012). McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet Journal of Rare Diseases, 7(Suppl 1), S4. doi:10.1186/1750-1172-7-s1-s4
-
Agopiantz, M., Journeau, P., Lebon-Labich, B., Sorlin, A., Cuny, T., Weryha, G., & Leheup, B. (2016). McCune–Albright syndrome, natural history, and multidisciplinary management in a series of 14 pediatric cases. Annales d’Endocrinologie, 77(1), 7–13.doi: 10.1016/ j.ando.2016. 01.002
-
Lee, J., FitzGibbon, E., Chen, Y., Kim, H., Lustig, L., Akintoye, S., Kaban, L. (2012). Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet Journal of Rare Diseases, 7(Suppl 1), S2.doi:10.1186/1750-1172-7-s1-s2
-
Bousson, V., Rey-Jouvin, C., Laredo,J.-D., Le Merrer, M., Martin-Duverneuil, N., Feydy, A., Orcel, P. (2014). Fibrous dysplasia and McCune–Albright syndrome: Imaging for positive and differential diagnoses, prognosis, and follow-up guidelines. European Journal of Radiology, 83(10), 1828–1842.doi: 10.1016/j.ejrad.2014.06.012
-
Leong, L. T. Y., & Ming, B. J. C. C. (2015). Craniofacial Fibrous Dysplasia Involving the Orbit. Asia-Pacific Journal of Ophthalmology, 4(3), 151–154.doi:10.1097/apo.0000000000000043
-
Denadai, R., Raposo-Amaral, C. A., Marques, F. F., Ghizoni, E., Buzzo, C. L., & Raposo-Amaral, C. E. (2016). Strategies for the Optimal Individualized Surgical Management of Craniofacial Fibrous Dysplasia. Annals of Plastic Surgery, 77(2), 195–200. doi:10.1097/sap.0000000000000640
-
Lecumberri, B., Pozo-Kreilinger, J. J., Esteban, I., Gomes, M., Royo, A., Gómez de la Riva, Á., & Pérez de Nanclares, G. (2018). Head and neck manifestations of an undiagnosed McCune-Albright syndrome: clinicopathological description and literature review. Virchow Archiv.doi:10.1007/s00428-018-2396-z.
-
Yew, L. (2015). Craniofacial Fibrous Dysplasia Involving the Orbit: A Case Report and Literature Review. Asia-Pacific Journal of Ophthalmology, (Number 3,), 151,152,153,154.
-
Hernández L., Espinoza A., Méndez V., Nishimura E., Mercado M. Síndrome de McCune Albright: características clínicas en una población pediátrica y adulta. Revista de Endocrinología y Nutrición. Vol 20. N.1 enero-marzo 2012
-
Deepthi, A. (2016). Fibrous dysplasia of the craniofacial region - A brief understanding of the disease. Journal of Advanced Clinical & Research Insights, (Vol. 3:6), 205–208.
-
Ballesteros, A. (2015). síndrome de mccune-albright: reporte de caso y revisión de la literatura. rev.medica.sanitas, (volumen 18 • no. 4), 236-239.
-
Guillén, L. (2012). Pubertad precoz periférica: fundamentos clínicos y diagnóstico-terapéuticos. Anales de Pediatría, (76), 229.e1---229.e10,
-
Florentín, Cynthia. (2014). McCune-Albright syndrome: a Case Report. Pediatr. (Asunción), (41), 139-1
-
Uribe González Guillermo, Sigler Morales Luis. Síndrome de McCune-Albright en un adolescente. Informe de un paciente. Cir. gen [revista en la Internet]. 2017 Mar [citado 2021 Abr 26]; 39(1): 37-40. Disponible en: http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S1405-0099201... Epub 26-Mar-2020.
-
Lovy A, Dowdell J, Keswani A, Koehler S, Kim J, Weinfeld S, et al. Nonoperative versus operative treatment of displaced ankle fractures in diabetics. Foot Ankle Int 2017; 38(3):255-60. DOI: 10.1177/1071100716678796.
-
Rienzi Tomás, Silveri Claudio, Risso Mariana, Mendoza Beatriz, Bianchi Gottardo. Displasia fibrosa poliostótica - síndrome de McCune-Albright: relato de um caso. Rev. Méd. Urug. [Internet]. 2021 Mar [citado 2021 Abr 26]; 37 (1): e701. Disponible en: http://www.scielo.edu.uy/scielo.php?script=sci_arttext&pid=S1688-0390202... Epub 01-Mar-2021. http://dx.doi.org/10.29193/rmu.37.1.12.
-
Santini-Araujo E, Kalil R, Bertoni F, Park Y. Tumors, and tumor-like lesions of bone. Basel: Springer Nature, 2020.
-
Brenner, T. C. M., Herter, L. D., & Kopacek, C. Síndrome de McCune-Albright: serie de casos. Rev Soc Arg. Ginecol Inf. Juv. Volumen 27 - Número 2 - 2020
_________________________________________________________
Disclosures
The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.
Author Contributions
Conception and design: All the authors. Drafting the article: Espín G, Torres A. Critically revising the article: Torres A, Espín G. Reviewed submitted version of the manuscript: Torres A. Approved the final version of the manuscript on behalf of all authors: Torres A.
Correspondence
Alicia Fernanda Torres Merino. Department of Pediatric Neurosurgery of the Baca Ortiz Pediatric Hospital. 6th floor, 6 December, and Colón Avenue. Quito, Ecuador. 15003. E-mail: alifertorres@hotmail.com, alifertorresme@gmail.com

